分析吡哆醇依赖性癫痫(PDE)患儿的临床及遗传学特点,建立尿液哌啶酸的检测方法,分析PDE患儿吡哆醇治疗过程中尿液哌啶酸的浓度。
对2012年4月至2015年9月在北京大学第一医院儿科确诊的12例PDE患儿(男8例、女4例)的临床表现、诊治过程、脑电图及神经影像学资料进行回顾性分析;采用Sanger测序或靶向捕获二代测序技术进行ALDH7A1基因检测;采用气相色谱-质谱(GC-MS)方法测定PDE患儿的尿液哌啶酸浓度,并检测非PDE患儿的尿液哌啶酸作为正常对照。对照组为2015年11月至2016年1月于北京大学第一医院出生的新生儿或因晕厥等原因(不影响哌啶酸代谢)就诊的儿童共58例,其中≤6月龄组25例(男14例、女11例),6月龄组33例(男14例、女19例)。哌啶酸浓度的组间比较采用两独立样本的t检验或Mann-Whitney检验;相关性分析采用Pearson或Spearman检验。
12例PDE患儿中7例母亲妊娠期或出生史异常,起病年龄为生后5 h~5个月,8例于生后10 d内起病。经过15 d~20个月的延迟诊断,患儿的癫痫发作最终均被吡哆醇单药控制,其中10例发作控制时吡哆醇剂量高于10.0 mg/(kg·d),另2例分别为8.5和2.5 mg/(kg·d)。发作间期脑电图显示10例有非特异性异常,2例吡哆醇用药前后均正常;头颅磁共振成像显示7例为非特异性异常,5例正常。患儿均通过ALDH7A1基因检测确诊,共发现15种突变位点,其中4种为国际尚未报道的新突变。6例携带IVS11+1GA突变,出现率25%(6/24)。至末次随访,11例患儿有不同程度的智力、运动发育落后,其中4例重度落后者出生史均有异常;1例未发现明显的发育异常。正常对照组中,≤6月龄组尿液哌啶酸浓度明显高于6月龄组[8.47(0.46~35.33)比0.66(0.12~3.52)mmol/mol肌酐,Z=-5.464,P0.01]。12例PDE患儿检测时年龄均6月龄,尿液哌啶酸浓度0.14~4.08 mmol/mol肌酐(仅有1例浓度稍高于正常值上限),与正常对照6月龄组差异无统计学意义(Z=-0.655,P0.05),与吡哆醇初始、末次随访维持剂量无相关性(r=0.418、0.166,P=0.176、0.697)。
PDE患儿多在新生儿早期起病。IVS11+1GA突变出现率较高,可能为我国PDE患儿的热点突变。多数患儿癫痫发作控制后遗留不同程度的智力、运动发育落后,出生史异常者提示预后较差。未发现诊断延迟时间与智力、运动发育情况之间存在相关性。尿液哌啶酸浓度经吡哆醇长期维持治疗后可能恢复正常。
吡哆醇依赖性癫痫(pyridoxine dependent epilepsy,PDE,OMIM 266100)由Hunt等[]在1954年首次报道,是一种少见的常染色体隐性遗传病,特征为新生儿期或婴儿早期出现难以控制的癫痫发作,且抗癫痫药无显著疗效,大剂量吡哆醇可完全控制发作[]。2006年,ALDH7A1基因被确定为PDE的致病基因[]。ALDH7A1基因编码α-氨基己二酸半醛(α-AASA)脱氢酶,参与体内赖氨酸的分解代谢,该基因突变会引起α-AASA累积,后者在体内与Δ1-四氢吡啶-6-羧酸(P6C)处于自发平衡状态,导致P6C继发性累积,并进一步引起体内哌啶酸累积[]。因此,PDE患者体内α-AASA、P6C及哌啶酸浓度升高。2012至2015年在北京大学第一医院儿科通过ALDH7A1基因分析共确诊12例PDE患儿,并建立气相色谱-质谱(GC-MS)检测尿液哌啶酸浓度的方法,对患儿吡哆醇治疗过程中哌啶酸浓度进行检测,现回顾性总结PDE的临床及遗传学特点,并分析哌啶酸浓度与吡哆醇维持剂量、智力及运动和发育情况之间的关系。
2012年4月至2015年9月在北京大学第一医院儿科确诊的PDE患儿12例(男8例、女4例)。入组标准:(1)新生儿期或婴儿早期出现癫痫发作;(2)癫痫发作对抗癫痫药耐受;(3)大剂量吡哆醇(维生素B6)单药完全控制发作;(4)基因检测明确ALDH7A1基因突变。本研究获得了北京大学第一医院临床研究伦理委员会的批准(2005年第04号),患儿家长均知情同意。
正常对照组为2015年11月至2016年1月于北京大学第一医院出生的新生儿或因晕厥等原因(不影响哌啶酸代谢)就诊于我院的儿童共58例。依据体内哌啶酸的年龄依赖性特点,将正常对照组分为2个年龄组(≤6月龄组,6月龄组)检测其尿液哌啶酸浓度作为正常对照[]。≤6月龄组25例(男14例、女11例),6月龄组33例(男14例、女19例)。
回顾性分析患儿的临床表现、诊治过程及视频脑电图(VEEG)、头颅磁共振成像(MRI)结果。
采用Sanger测序或靶向捕获二代测序技术对PDE患儿进行ALDH7A1单个基因或癫痫相关基因包(含ALDH7A1基因)检测及分析[]。
委托北京摩尔美康分离检测技术有限公司完成,采用GC-MS分析方法检测。标准品、萃取试剂、衍生化试剂均购自北京百灵威科技有限公司。(1)将350 μl尿液移入制备小瓶,加入150 μl甲醇溶液,充分混匀后,高速离心12 000 r/min(离心半径6 cm)×10 min;移取上清液400 μl,加入1 ml乙酸乙酯萃取试剂进行液液萃取,将萃取后溶液常温下氮气吹干;加入100 μl BSTFA衍生化试剂,80 ℃温育30 min对样品进行衍生化;最后将样品移入样品瓶进行GC/MS分析。(2)GC/MS联用仪分析条件:①气相:色谱柱Rtx-5MS 30 m×0.25 μm×0.25 mm;进样口温度280 ℃;进样方式为分流(1∶10);流量控制模式为压力(76.9 kPa)控制;线 cm/s;升温程序为100 ℃→10 ℃/min→300 ℃(10 min)。②质谱:离子源温度200 ℃;接口温度:280 ℃;溶剂切除时间为2.5 min。(3)以标准品所制标准曲线.辅助检查:
完善血液生化检查、血液氨基酸、尿液有机酸筛查等,进行头颅MRI检查及智力及评估(Gesell发育量表、临床粗略智力评估)。
通过门诊随诊或电话随访方式获得近期资料,主要包括发作情况、吡哆醇维持剂量、智力及运动发育情况等,随访时间为5个月至3.5年。
用SPSS 20.0软件进行统计学处理,采用Shapiro-Wilk检验方法对资料进行正态性检验;正态分布的计量资料用±
表示,非正态分布的采用中位值(范围)表示,组间比较采用两独立样本的t检验或Mann-Whitney检验,相关分析采用Pearson或Spearman检验。P0.05为差异有统计学意义。
12例患儿均经临床及基因检测确诊为PDE()。3例有生后窒息,2例有羊水污染,1例妊娠期有先兆流产,1例妊娠期有异常胎动,另5例出生史正常。患儿均为新生儿期或婴儿期起病,起病年龄为生后5 h~5个月,8例于生后10 d内出现癫痫发作。患儿均为局灶性发作,表现为双眼凝视或斜视,一侧或双侧肢体抽搐,伴或不伴口唇发绀。经过15 d~20个月的延迟诊断后,12例患儿的癫痫发作最终均被吡哆醇单药控制,其中10例发作控制时吡哆醇剂量高于10.0 mg/(kg·d),另2例(例4、11)分别为8.5和2.5 mg/(kg·d)。VEEG检查结果显示10例发作间期为局灶性或多灶性尖波、尖慢波发放,2例在吡哆醇治疗前后经多次VEEG检查结果均正常。头颅MRI检查7例显示多种非特异性异常,包括白质信号异常、颞区蛛网膜下腔增宽、胼胝体发育不良、脑室扩大和脑发育不良等,5例未发现异常。所有患儿血液生化、血液氨基酸、尿液有机酸筛查等均未见异常。
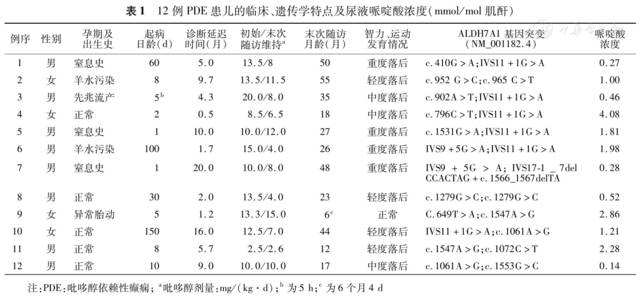
ALDH7A1基因分析显示11例为复合杂合突变;1例为纯合突变。检测患儿父母基因型确定上述突变均分别遗传自父母,符合PDE的常染色体隐性遗传特性。共发现15种不同的突变位点(),其中4种(c.649TA、c.1061AG、c.1072CT、c.1553GC)为国际未报道的新突变,IVS11+1GA突变出现于6例患儿中。
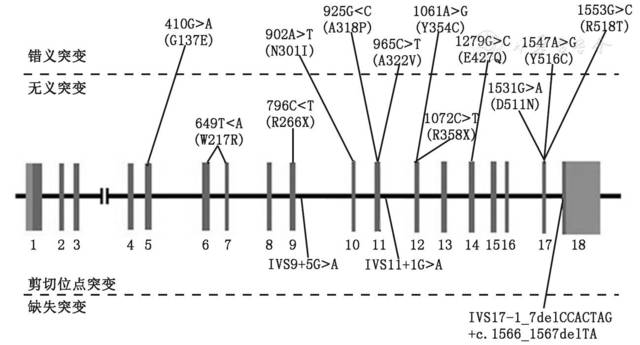
采用Sanger测序或目的基因靶向捕获二代测序技术获得12例PDE患儿的ALDH7A1基因突变位点,其中Y354C、E427Q、Y516C、IVS9+5GA、IVS11+1GA出现次数≥212例患儿分别随访5个月~3.5年。至末次随访,患儿年龄为6个月4 d~4岁7个月,吡哆醇维持剂量2.6~15.0 mg/(kg·d),所有患儿的癫痫发作均被完全控制。11例有不同程度的智力、运动发育落后,其中4例轻度落后、3例中度落后、4例重度落后,例9年龄尚小未发现明显的发育异常。
=-5.464,P0.01]。12例PDE患儿进行尿液哌啶酸浓度检测时年龄均6月龄,结果为0.14~4.08 mmol/mol肌酐(),与正常对照6月龄组差异无统计学意义(Z=-0.655,P=0.513)。PDE患儿尿液哌啶酸浓度与吡哆醇初始剂量、末次随访维持剂量之间均无相关性(r=0.418、0.166,P=0.176、0.607)。PDE是少见的常染色体隐性遗传病,国际共报道200余例,有关发病率的报道较少且差异很大,为1∶20 000~1∶600 000[]。目前PDE的诊断首先依靠临床疑诊,有条件的实验室可检测生化标志物进一步提示诊断,最后基因检测确诊。由于此病少见,临床医生诊断经验不足,常导致漏诊而延误治疗。本组12例中7例妊娠期或出生史异常,且1例妊娠期曾有异常胎动,提示宫内发作的可能性[]。8例患儿生后10 d内出现癫痫发作,但有1例生后5个月起病,更有报道指出起病年龄可晚至3岁[]。既往研究报道PDE患儿发作形式多变,包括局灶性发作、痉挛发作、肌阵挛发作、强直-阵挛发作、失张力发作,甚至癫痫持续状态等[],本组患儿均为局灶性发作,可能因病例数尚少,不能全面反映PDE的发作类型;既往报道的PDE病例多为临床拟诊,未经基因检测确诊,有可能将部分吡哆醇反应性癫痫患儿误诊为PDE所致,如吡哆醇反应性婴儿痉挛[]。本组中VEEG及MRI检查结果均未发现可提示诊断的特异性改变。本研究发现的15种ALDH7A1突变位点中,4种为国际未报道的新突变,另有5种已由本课题组首次报道[]。ALDH7A1基因共包含18个外显子,突变主要集中于第11、12、14、17号外显子上,其中第11内含子IVS11+1GA突变出现率较高(25%),可能为我国PDE患儿的热点突变[],提示在进行基因筛查时应重点考虑这些区域。
至末次随访,所有患儿的癫痫发作均被吡哆醇单药控制,但11例出现不同程度的智力、运动发育落后,且4例重度落后患儿出生史均有异常,提示此类患儿预后相对较差。但是,出生史正常的患儿预后存在个体差异性;不同患儿延迟诊断时间不同且吡哆醇维持剂量也不尽相同,并未发现诊断延迟时间与智力、运动发育情况之间的明确相关性,早治疗并不能确保预后好。PDE预后可能主要受ALDH7A1基因突变位点的影响[],但本组中1例携带E427Q纯合突变的患儿智力、运动发育仅为轻度落后,而据报道,E427Q突变会影响NAD+辅酶结合或催化区域,从而使酶活性完全丧失[],因此携带此突变的患儿预后较差。本研究及国外的研究提示本病预后除受基因型影响外,其他因素如起病年龄、饮食中赖氨酸的摄入量、合成及分解代谢差异、其他环境因素等对预后也有一定的影响[]。
ALDH7A1基因突变可导致血液、尿液、脑脊液中的α-AASA、P6C和哌啶酸浓度升高,三者均可作为诊断PDE的生化标志物[]。其中,α-AASA是α-AASA脱氢酶的直接底物,在基因突变后最先被影响,并与P6C处于自发平衡状态,故PDE患者体内α-AASA和P6C浓度远高于哌啶酸[]。由于α-AASA和P6C稳定性差,在室温下极易降解[],检测困难且难以精确定量,较难应用于临床。因此,尽管哌啶酸的特异性相对较低[],但其稳定性好、灵敏度高,易于临床普及、应用。国际上仅有美国、荷兰、奥地利等少数国家可进行PDE生化标志物的检测[]。考虑到尿液相对易于获得且无创,我们建立了通过GC-MS检测尿液哌啶酸浓度的方法。根据哌啶酸具有年龄依赖性的特点,分组收集作为正常对照;检测结果与已报道的GC-MS方法测得的正常范围(mmol/mol肌酐:≤6个月者为0.55~24.1;6个月者为0.01~1.54[])相近,证明所建方法可靠。因α-AASA的化学本质是活性半醛,可参与细胞内的多种化学反应,继而影响多条代谢通路,PDE患儿体内α-AASA蓄积可能对脑组织产生直接的毒性损害[],尿α-AASA浓度高者临床表型更严重[],而哌啶酸累积继发于α-AASA蓄积,因此,尿液哌啶酸浓度可能一定程度上反映患儿预后;有报道,尿液哌啶酸浓度在治疗之前可高至正常高限的17倍,长期应用吡哆醇治疗后可明显降低甚至恢复正常[],因此,尿液哌啶酸水平可能有助于调整吡哆醇维持剂量。本研究进行哌啶酸检测时,12例PDE患儿均为吡哆醇单药治疗,且吡哆醇维持剂量与末次随访时一致,仅有1例稍高于上限,但并未发现此例吡哆醇剂量过低或智力、运动发育情况更差。PDE患儿与6月龄正常对照组间哌啶酸浓度无明显差异,推测可能与本组PDE患儿均经数月至数年的吡哆醇治疗,尿液哌啶酸浓度多数已恢复正常有关,较为遗憾的是由于哌啶酸检测方法刚刚建立,本组患儿均未能在吡哆醇治疗前被证实尿液哌啶酸水平高于正常。研究提示PDE患儿个体间异常增高的尿液哌啶酸浓度差异较大,考虑哌啶酸浓度更可能由所携带突变位点不同决定[]。但当个体间哌啶酸浓度在正常值范围内,此时数值上的差异不具有病理性意义。
本研究通过建立尿液哌啶酸检测方法,使我国PDE的生化标志物的快速筛查成为可能。未来对新诊断的PDE患儿可测定吡哆醇治疗前、治疗后不同病程阶段的尿液哌啶酸水平,以助于PDE的早期诊断,并可指导临床吡哆醇长期用药。
《中华儿科杂志》微信公众号由《中华儿科杂志》编辑部、易慧睿思品牌咨询有限公司共同运营,是中华医学会杂志社旗下所属《中华儿科杂志》的唯一官方微信平台
”,联系我们。注:单期购买时,请在出版日前15天下单,逾期商品将下架。出版日可在“
